Les Organismes Notifiés en Europe : toujours trop peu face au défi MDR/IVDR

Índice de contenidos
Index of contents
Index du contenu
Inhaltsverzeichnis
Indice dei contenuti
- Introduction : le paradoxe d’une demande croissante face à une offre limitée
- Rappel réglementaire : MDR, IVDR et rôle des Organismes Notifiés
- Quelques chiffres clés
- Le point de rupture : pourquoi le manque d’ON est un risque systémique
- Projections : à quoi s’attendre d’ici 2028 ?
- Impacts pour les fabricants et les PRRC / QARA
- Que peut-on faire pour améliorer la situation ?
- Conclusion : le compte à rebours est lancé
Introduction : le paradoxe d’une demande croissante face à une offre limitée
Avec l’entrée en vigueur du MDR et de l’IVDR, l’ensemble des fabricants de dispositifs médicaux devront passer par des Organismes Notifiés (ON). Chaque dispositif ou famille de dispositifs devra être certifié conforme au règlement pour pouvoir être mis sur le marché européen. Pourtant, le nombre de ces organismes désignés reste insuffisant pour absorber le flux de demandes.
Rappel réglementaire : MDR, IVDR et rôle des Organismes Notifiés
— Le règlement (UE) 2017/745 (MDR) entré en application le 26 mai 2021 remplace les anciennes directives.
— Le règlement (UE) 2017/746 (IVDR) pour les dispositifs in vitro entre en application en 2022.
— Les Organismes Notifiés sont les entités habilitées à réaliser les évaluations de conformité, notamment pour les dispositifs de classe plus élevée, évaluer la documentation technique, auditer les systèmes qualité, etc.
Quelques chiffres clés
Selon MedTech Europe, le secteur des Dispositifs Médicaux et In-Vitro en Europe représente :
- Environ 38,000 entreprises
- Plus de 500,000 dispositifs en circulation en 2025 (dont à minima 80% sont encore sous les Directives européennes 93/42/CEE et la 98/79/CE)
La transition vers le MDR & IVDR
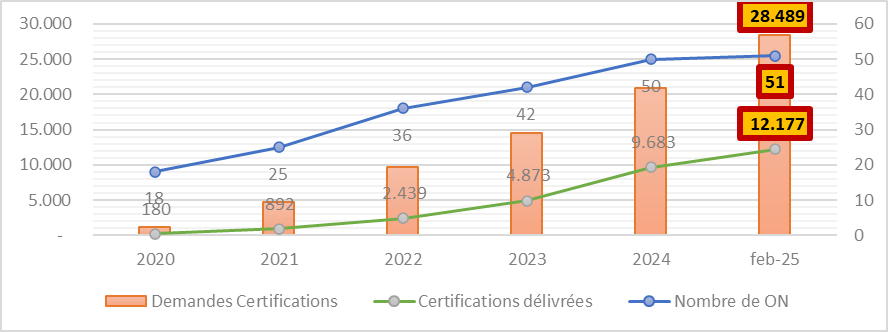
En février 2025, selon les données mis en lumière par la Commission Européenne, les 51 ON officiellement désignés doivent traiter les demandes de certification dont le nombre total se trouve à hauteur de 28,500. À la même date plus de 12,000 certificats ont été accordés, soit 43%.
Ce qu’il faut également savoir :
- Un dossier mets en moyenne entre 13 et 18 mois pour obtenir une certification
- Les dossiers soumis incomplets rallongent ce délai
- Les dossiers peuvent être refusés et doivent donc être représentés
Le point de rupture : pourquoi le manque d’ON est un risque systémique
— Goulots d’étranglement : les ON existants croulent sous les demandes, leurs ressources (expertise, auditeurs) sont limitées.
— Complexité accrue des règlementations : le MDR impose des exigences plus strictes (clinique, surveillance post-market, documentation renforcée) que sous les directives, ce qui alourdit la charge de travail par dossier.
— Dossiers incomplets ou mal préparés : implique un perte de temps précieux pour les ON et les fabricants
— Risque de pénurie de dispositifs sur le marché européen si les fabricants ne parviennent pas à obtenir leur certification à temps (retard d’accès au marché). Une des motivations de l’extension des périodes transitoires est précisément d’éviter ces ruptures.
— La pression temporelle monte : avec la date butoir de certification complète prévue d’ici 2028, le temps disponible pour les transitions est de plus en plus réduit.
Projections : à quoi s’attendre d’ici 2028 ?
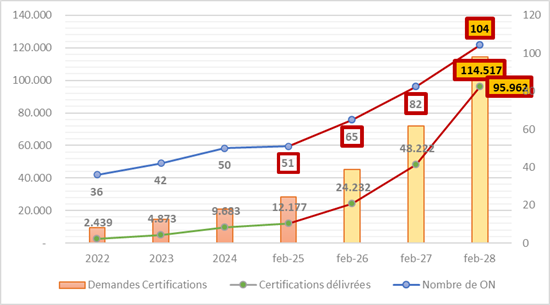
Projection 1 : En se basant sur le rythme de désignation d’ON entre 2022 et 2025, on pourrait espérer passer de 51 actuellement à environ 85 ON d’ici fin 2027 et plus de 100 en 2028. Mais cela dépend fortement de la volonté politique, des investissements nationaux, et des capacités d’accréditation.
Projection 2 : Toujours en se basant sur les évolutions depuis 2022, le nombre de demandes atteindrait au moins 114,000. Ce chiffre n’est cependant pas pertinent et devrait être donc considérablement plus important car environ 400,000 dispositifs sur les 500,000 actuellement en circulation doivent passer sous le règlement. Et ce malgré les regroupements possibles par « famille de dispositifs » (sinon 1 dispositif = 1 dossier).
Projection 3 : Selon le rythme actuel de certifications, on peut estimer que 95,000 certificats délivrés le seront en 2028. Ce n’est même pas au niveau des 114,000 dossiers possibles dans le meilleurs des cas.
Résultats des projections : Si le nombre de 104 ON est effectivement atteint d’ici 2028 et que le nombre de certificats délivrés l’est également, cela représente 919 certifications par ON. Est-ce viable ?
Impacts pour les fabricants et les PRRC / QARA
Du côté des personnes en charges des Affaires Réglementaires la pression est palpable. Les différentes contraintes mentionnés avant (temps, budget, exigences règlementaires, etc.) leur rendent la tâche complexe car :
— La sélection d’un ON devient un défi stratégique : disponibilité, délais, compétence, tarification, spécialisation par type de dispositif.
— Une planification proactive indispensable : commencer tôt la préparation des dossiers, capitaliser sur les audits préliminaires, s’assurer que la documentation est complète et conforme.
— Il existe un risque de décalage sur les lancements : un produit prêt doit attendre l’évaluation du dossier par l’ON pour entrer sur le marché.
— Ils doivent parfois avoir recours à des ON à l’étranger (coûts, logistique).
— Ils savent l’importance de la stabilité du partenariat avec l’ON pour prévoir les audits de surveillance, les renouvellements, les modifications de portée, etc.
Malgré les efforts des différentes parties prenantes, la complexité des exigences des règlement et le manque criant d’ON rendent la tâche difficile. Les acteurs du secteurs, dont les ON eux-mêmes, interpellent régulièrement les autorités pour pointer les lacunes structurelles et alerter sur l’effet d’étranglement et de pénurie.
Que peut-on faire pour améliorer la situation ?
Dans le contexte évoqué avant il n’existe pas de solution miracle. Si l’Europe veut éviter une grave pénurie de dispositifs médicaux, elle doit agir rapidement notamment en étudiant les proposition suivantes :
— Encourager les États membres à promouvoir et soutenir la désignation d’ON supplémentaires.
— Harmoniser et simplifier certaines étapes du processus d’évaluation pour alléger la charge administrative. (En revoyant certaines exigences ?)
— Renforcer la formation d’auditeurs experts MDR/IVDR pour augmenter le vivier de compétences disponibles.
— Assurer la qualité documentaire des fabricants pour éviter les itérations inutiles. (Avec des explications claires et précises de ce qui est requis)
— Établir un délai légal de réponse pour les ON ?
— Favoriser la collaboration entre ON, agences nationales et associations industrielles pour mieux répartir les charges.
Conclusion : le compte à rebours est lancé
Le déficit d’Organismes Notifiés en Europe reste l’un des freins majeurs à une transition fluide vers un marché pleinement conforme aux règlementations MDR/IVDR. Pour les fabricants comme pour les PRRC / QARA, cette réalité impose une stratégie rigoureuse, anticipée et résiliente. Si la désignation de nouveaux ON progresse, elle doit être assortie d’une montée en capacité réelle et d’une efficacité accrue pour éviter le risque d’engorgement généralisé d’ici 2028.
Derniers chiffres mis à jour Octobre 2025 :
- Organismes notifiés pour le Règlement EU 2017/745 Dispositifs Médicaux : 51
- Organismes notifiés pour le Règlement EU 2017/746 Dispositifs In Vitro : 19
Sources et références
- Commission Européenne – Study supporting the monitoring of the availability of medical devices on the EU market, 2025.
https://health.ec.europa.eu/document/download/59b9d90e-be42-4895-9f6f-bec35138bb0a_en?filename=md_nb_survey_ - MedTech Europe – The European Medical Technology Industry in figures, 2025.
- Commission Européenne – NANDO ( New Approach Notified and Designated Organisations ) Information System.
https://webgate.ec.europa.eu/single-market-compliance-space/notified-bodies
Ces articles pourraient vous intéresser :



Issu d’une formation en Marketing et Commerce International, Alex a toujours exprimé un attrait pour les langues et intérêt pour les différentes cultures. Originaire de Bretagne en France, il a vécu en Irlande et au Mexique avant de repasser un temps par la France puis s’établir définitivement en Espagne. Il est Chief Growth Officer au sein d'AbroadLink.
Ajouter un commentaire